
High-resolution Mapping of Linear Antibody Epitopes Using Ultrahigh-density Peptide Microarrays* 
Associated Data
- Supplementary Materials
- Supplementary informationsupp_11_12_1790__index.html (1021 bytes)
Abstract
Antibodies empower numerous important scientific, clinical, diagnostic, and industrial applications. Ideally, the epitope(s) targeted by an antibody should be identified and characterized, thereby establishing antibody reactivity, highlighting possible cross-reactivities, and perhaps even warning against unwanted (e.g. autoimmune) reactivities. Antibodies target proteins as either conformational or linear epitopes. The latter are typically probed with peptides, but the cost of peptide screening programs tends to prohibit comprehensive specificity analysis. To perform high-throughput, high-resolution mapping of linear antibody epitopes, we have used ultrahigh-density peptide microarrays generating several hundred thousand different peptides per array. Using exhaustive length and substitution analysis, we have successfully examined the specificity of a panel of polyclonal antibodies raised against linear epitopes of the human proteome and obtained very detailed descriptions of the involved specificities. The epitopes identified ranged from 4 to 12 amino acids in size. In general, the antibodies were of exquisite specificity, frequently disallowing even single conservative substitutions. In several cases, multiple distinct epitopes could be identified for the same target protein, suggesting an efficient approach to the generation of paired antibodies. Two alternative epitope mapping approaches identified similar, although not necessarily identical, epitopes. These results show that ultrahigh-density peptide microarrays can be used for linear epitope mapping. With an upper theoretical limit of 2,000,000 individual peptides per array, these peptide microarrays may even be used for a systematic validation of antibodies at the proteomic level.
The immune system is endowed with a highly diverse repertoire of antibodies capable of targeting virtually any molecular structure. As specific affinity reagents, antibodies have become indispensable tools with a wide range of scientific and diagnostic applications (1, 2). Thus, antibodies are the main priority of several recent initiatives such as the Human Protein Atlas (3) and the ProteomeBinders consortium (4, 5) and of efforts to generate antibodies against cancer-related targets (6, 7), all of which aim to systematically generate affinity reagents, thereby facilitating the study of proteins and their role in biology and disease. As therapeutic agents, monoclonal antibodies have emerged as essential drugs with a wide range of clinical applications, making monoclonal antibodies one of the highest priorities of the pharmaceutical industry (8–11). The efficiency, accuracy, and safety of these antibody-mediated applications depend crucially on the selected antibodies being directed against the intended, and not against any unintended, target structure(s) (12). Specificity, the quintessential characteristic of an antibody, is therefore not only of scientific interest, but also of considerable practical importance.
For any antibody-based application, the establishment of specificity constitutes an important aspect of the validation process. Traditionally, the specificity of an antibody is examined in one or more in vitro assays (ELISA, Western blot, immunohistochemistry, flow cytometry, surface plasmon resonance, and many more (12–14)). Ideally, the entire epitope space should be examined; however, it is rarely possible to test more than a minor and ostensibly relevant part of the epitope space. What is relevant depends on the intended use; thus, the same antibody might exhibit sufficient and relevant specificity in one, but not in another, application (15). An important aspect of validating the specificity of an antibody is to determine the structure of the epitope that the antibody interacts with (12). Ideally, one would like to determine the three-dimensional structure of the binding complex using x-ray crystallography (16–18) or NMR1; however, such efforts are laborious and tend to have a low success rate and throughput. Many other epitope mapping approaches, such as fragmentation (19) or deuterium exchange in the presence or absence of antibody (20), directed mutagenesis, recombinant expression (including arrayed in situ cell-free translation approaches (20, 21)) of protein and peptide arrays, etc., have been suggested (12). Despite this plethora of methods, exact epitope information is lacking for the vast majority of antibodies used in life science research, and there is a significant need for simple and rapid methods to map epitopes. The availability of such methods would also support the selection of paired antibodies that each bind to separate parts of an antigen, thereby allowing one antibody to validate the results of another (12, 22).
Proteins constitute important immune targets, and many of the methods used to address antibody specificity are tailored for protein antigens. Traditionally, protein epitopes have been divided into discontinuous/conformational epitopes, which require that the native protein structure be intact, or continuous/linear epitopes, which may be represented by consecutive overlapping synthetic peptides encompassing the complete primary structure of the target antigen (15). The mapping resolution of linear epitopes depends on the peptide length, the overlap chosen for the initial epitope location, and the scale of the subsequent fine specificity analysis (e.g. N- and C-terminal truncations; amino acid scans; random single, double, or triple substitutions; etc.). The number of peptides required can be substantial, making the cost of peptides and the logistics of handling large panels of peptides a serious impediment of the in-depth characterization of linear epitopes. Most standard peptide synthesis equipment can synthesize only up to a few hundred single peptides simultaneously, although lately up to 8000 peptides have been synthesized in parallel on a cellulose membrane (23–25) using the SPOTTM technique. In addition to performing assays directly on the membrane (26), such peptides can be released and transferred onto glass slides using additional robotics and printing techniques (25). As alternatives to synthetic peptides, phages (27), bacteria (28), and yeast cells (29) have been used to express libraries of fragmented antigens (27) or of combinatorial peptides (30). These methods can potentially generate millions of peptides covering entire protein antigens, and they may, at least in some cases, mimic conformational epitopes (15, 31–33). Major drawbacks of these methods include the lack of control of the exact peptide sequences expressed and the need for separate sequencing of positive clones. None of these drawbacks are encountered with peptide microarrays.
Here, we present the first report on the feasibility of using ultrahigh-density peptide microarrays to address antibody specificities in casu mapping the fine specificity of polyclonal antibodies raised against linear protein epitopes. This allowed a fast and exhaustive analysis of the length requirements and a detailed analysis of the fine specificity of these antibodies. We suggest that specificity analysis of linear epitopes using ultrahigh-density peptide microarrays addressing the entire human proteome is within reach.
EXPERIMENTAL PROCEDURES
Derivatization of Synthesis Slides
Microscope slides (Nexterion E; Schott AG, Jena, Germany) for synthesis of the arrays were derivatized via incubation with 1 g/l bovine serum albumin in 0.5 m N-methylmorpholine (NMM)/acetate pH 8.5 for 3 h at room temperature. The slides were washed in water, N-methylpyrrolidone (NMP), and dichloromethane (DCM) and stored dry until use. Synthesis of the microarrays was performed directly on the BSA-coated slides using the epsilon amino groups of sterically exposed lysines as the starting point.
Synthesis
Peptide arrays were synthesized by Schafer-N (Copenhagen, Denmark) using a maskless photolithographic technique (34) in which 365 nm light with an energy density of ca. 20 mW/cm2 was projected onto 3′-nitrophenylpropyloxycarbonyl (NPPOC)-photoprotected (35, 36) amino groups on a glass surface in patterns corresponding to the synthesis fields. Details of the technique will be published elsewhere, but briefly, the patterns were generated using digital micromirrors and projected onto the synthesis surface using UV-imaging optics (supplemental Fig. S1A). In each layer of amino acids, the relevant amino acids were coupled successively to predefined fields after UV-induced removal (in 1 mdiisopropylethylamine (DIEA) in NMP) of the photoprotection groups in those fields. The couplings were made using standard Fmoc-amino acids activated with O-benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate/DIEA in NMP. After coupling of the last Fmoc-amino acid in each layer, all Fmoc-groups were removed in 20% piperidine in NMP and replaced by NPPOC groups (37) coupled as the chloroformate in DCM with 0.1 m DIEA. The procedure was then repeated until all amino acids had been added to the growing peptide chains (supplemental Fig. S1B). Final cleavage of side protection groups was performed in TFA:1,2-ethanedithiol:water 94:2:4 v/v/v for 2 h at room temperature.
Epitope Mapping Using Peptide Arrays
Primary rabbit polyclonal antibodies were diluted to a concentration of around 100 ng/ml in PBS-Tween. Deprotected slides were blocked and hydrated overnight in a mixture of 1 g/l bovine serum albumin and 0.1% v/v detergent (Tween 20) in PBS and incubated for 1 h at room temperature with relevant polyclonal anti–protein epitope signature tag (PrEST) rabbit antibodies as primary reagents. After washing, the slides were incubated for 1 h at room temperature with Alexa Fluor 488-labeled goat-anti-rabbit IgG (Invitrogen, Carlsbad, CA) as a secondary reagent. Images of the stained arrays were recorded using an MVX10 fluorescence microscope equipped with an XM10 cooled digital camera (both from Olympus, Ballerup, Denmark) and analyzed using the analysis program PepArray (Schafer-N, Copenhagen, Denmark). See the supplementary information for a brief description of the PepArray program.
Epitope Mapping Using Cell-surface Display
Mapping using cell-surface display was performed as described elsewhere (28). Briefly, gene fragments encoding the different antigens were amplified separately via PCR (4.8 ml pooled), and the products were sonicated to generate random fragments. These were blunt-ended and phosphorylated before ligation into the cell-surface expression vector pSCEM2 and transformed into Staphylococcus carnosus. Cell aliquots of about 10-fold coverage of the library were incubated with about 1 ng antibody in reaction volumes of 70 μl PBS-P. Cells were washed and fluorescently labeled with Alexa 488 secondary goat-anti-rabbit antibodies (Invitrogen, Carlsbad, CA) and Alexa 647 labeled albumin for expression normalization and then washed again ahead of analysis via FACS. Single cells expressing antibody-binding peptides were sorted, sequenced, and aligned back to the target protein sequence.
RESULTS
Ultrahigh-density Peptide Microarrays
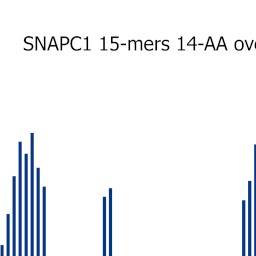
Peptide arrays were generated by a combined maskless photolithographic (34) and solid phase peptide synthesis strategy using a digital mirror device (1080P DMD (Digital Light Projections, Digital Light Innovations, Austin, TX) with 1920 × 1080 = 2,073,600 individually addressable micromirrors) to project 365 nm light onto NPPOC-photoprotected (35, 36) amino groups on a glass surface in patterns corresponding to the fields where the next amino acid extension should occur (supplemental Fig. S1A). Successively removing photoprotection groups extending the growing peptide chain with standard Fmoc-protected amino acids and exchanging the Fmoc-groups with NPPOC-groups after all extensions in a given layer (37) allowed individually predefined peptides to be built in each synthesis field (supplemental Fig. S1B). After synthesis of the peptide backbones, all side chain protection groups were removed via TFA treatment, leaving the peptides attached to the matrix through their C-terminals. Typically, each synthesis field was defined by a square measuring 2 × 2 (as in Fig. 1) or 3 × 3 mirrors. However, because synthesis fields defined by as few as one mirror could be discerned (Fig. 1), the maximum number of different synthetic peptides that can be realized with the current DMD device appears to be around 2,000,000 on a surface area of ∼2 cm2.




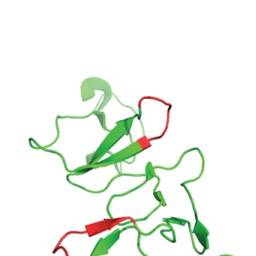
Epitope context and structure. Epitopes identified by the peptide microarray approach and reported in Fig. 4were mapped onto the known structure of the underlying proteins. Epitopes located on several different secondary structural elements, including parallel beta-sheets, loops, and helical regions, could be identified. In five of the seven cases shown here, several distinctly separated epitopes were identified.
DISCUSSION
Using a photolithographic approach, one of us has recently developed an ultrahigh-density peptide microarray technology theoretically capable of expressing up to 2,000,000 individual peptides on a ca. 2 cm2 area (41, 42). This has been achieved through a combination of previously reported advances in peptide microarray technology and chemistry. In a seminal 1991 study, Fodor et al. (43) used photo-masks and activated amino acids, which had been synthesized individually with a photolabile protection group, to generate a rather costly photolithographic principle for the synthesis of pre-addressable peptide microarrays. However, this technology was outcompeted by the cheaper and simpler SPOT principle of peptide array synthesis, which was introduced around the same time (44); for a historical overview, see Ref 45. Resurrecting photolithography as a principle of peptide microarray synthesis, Gao and co-workers (46, 47) used a DMD and photo-generated acids to effect light-directed peptide synthesis; however, this technology is limited by the need for physical barriers to confine the acid and prevent diffusion to unwanted areas, which also limits the peptide density that can be achieved. Recently, Li et al. (37) reported a chemical strategy for the in situ addition of photo-cleavable protection groups to a growing peptide chain, thus reestablishing a nondiffusable elongation principle. We have combined these advances, allowing a high-resolution DMD-driven photolithographic strategy without the need for photo-masks, physical barriers, or unique amino acid reagents (see supplemental Fig. S1), and basically allowing the use of standard solid-phase peptide synthesis reagents (40, 41).
Fig. 1 shows that this ultrahigh-density peptide microarray approach can achieve single mirror resolution and thus theoretically generate up to 2,000,000 peptides per microarray. By the same token, each of the 10 μm × 10 μm synthesis fields expresses very little peptide (estimated to be in the attomolar range). At this stage, it is not technically possible to address the quantity or quality of the peptides synthesized in each field (however, using all >2,000,000 mirrors to synthesize one and the same peptide, we have isolated sufficient material from a slide to ascertain that the intended peptide was indeed synthesized (data not shown)). A future hope would be that a high-sensitivity and label-free technology such as mass spectrometry could be adapted to validate the identity and purity of the peptides synthesized in each field, and potentially even be able to identify any reactant(s) offered to the peptide microarray. In the absence of such a technology, we have here resorted to a less direct, functional validation approach.
In this study, we have used an ultrahigh-density peptide microarray technology to map the location and fine specificity of a panel of polyclonal antibodies raised against short linear protein fragments uniquely representing human proteins (PrESTs). These antibodies were derived as part of the Human Protein Atlas initiative, which aims at generating specific antibodies against every protein of the human proteome (38). This initiative, together with other proteome-wide analysis initiatives, illustrates the need for new high-throughput technologies. Conventional solid phase peptide synthesis is obviously not able to provide the numbers of peptides needed to identify and validate proteome-wide reagents. Even array technologies based on pin synthesis or spotting approaches would be seriously challenged by these demands.
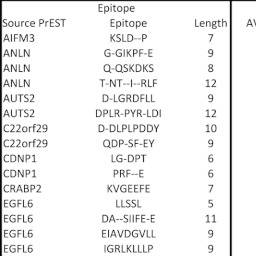
The ability to synthesize several hundred thousand peptides allowed us to address the specificity of 22 polyclonal antibodies using exhaustive and high-resolution length scans and SSA. This led to the identification of one or more epitopes for 20 of these 22 polyclonal antibody preparations. Five of the antibodies recognized only one epitope on their respective PrEST targets, whereas 15 of the antibodies recognized multiple discontinuous epitopes (up to seven epitopes per target). Our data confirm our previous report that antibodies within a polyclonal mixture can simultaneously be tested and used to identify linear peptide epitopes and that polyclonal antibodies, despite theoretically being able to target epitopes along the entire PrEST sequence, map to a few separate and distinct regions, suggesting that the majority of the target sequence is “epitope silent” (28, 39). Why some regions of these PrESTs remain epitope silent is not known. From a technical point of view, these epitope silent regions can be considered built-in negative experimental controls.
The length scans show the value of an exhaustive approach. As illustrated in Fig. 2B, some reactivity started being detectable at the level of 6-mer peptides, whereas others did not appear until at the level of 7-mer, 9-mer, or even longer peptides. This illustrates a fundamental problem in defining epitopes solely using overlapping peptides. Although short regions of strong reactivity probably represent dominant epitopes, this interpretation is confounded by the risk of the detected reaction being caused by two or more overlapping epitopes that might not have been resolved into individual epitopes. The closely positioned minimal epitopes EF-DPS and KLITSDVL illustrate this point. In this case, the minimal epitopes are sufficiently separated that each of them can be isolated and identified with short peptides; however, they would have been difficult to resolve if they had been more closely positioned.
One would be naturally inclined to compare the signal strengths of different peptide–antibody interactions and interpret them in terms of affinity. In this context, a word of caution is appropriate, because signal strength is determined by many factors. The relative contributions of these factors are not sufficiently controlled and/or known at this point. Thus, one should be careful when comparing different epitopes: a weak signal could theoretically be due to peptide synthesis failure, variations in peptide solvation, and/or the absence of high-affinity antibodies in reasonable concentrations. In this context, complete substitution analyses gave a very detailed, yet simple, description of the fine specificity of the epitope–antibody interactions and in many cases yielded highly significant results despite the weakness of the underlying signals. Thus, exhaustive SSAs followed by ANOVA and post hoc tests like Tukey’s LSD proved to be an efficient way to perform epitope calls and identify positions of selectivity. Fig. 4 illustrates how this statistical analysis in terms of epitope calling is superior to the mere recording of signal strength, which would have led to several otherwise clearly selective epitopes being discarded.
To our knowledge, this is one of the largest collections of fully substituted antibody epitope mappings reported. Some information on the biology of antibody recognition of linear epitopes can be extracted. The lengths of the epitopes were mostly 7 to 9 amino acids long (range: 4–12). In general, the epitopes contained a few very selective positions where the original amino acid was almost exclusively preferred and the signal dropped dramatically if the original amino acid was substituted with any other amino acid. In a few positions, the signal dropped less dramatically when conservative substitutions were made. In yet other positions, no significant contributions to the specificity could be detected. Thus, highly stringent, more relaxed, and nonselective positions could be intermingled as shown for the EF-DPS epitope. Our data show that polyclonal antibodies can be extremely selective peptide binders. We have previously examined the peptide binding specificity of MHC molecules, which have evolved specifically to present oligopeptides to T lymphocytes. In line with the requirement of MHC molecules to sample many different peptides, the specificity requirements of MHC molecules are much more relaxed. Structurally, MHC molecules achieve this broad specificity through extensive interactions with the peptide backbone. By inference, one could speculate that the highly selective peptide–antibody interactions are dominated by peptide side-chain interactions.
As alluded to previously, there are some important limitations to the ability of the current peptide microarray technology to address protein-specific antibody epitopes, as peptides do not readily represent more complex structures such as discontinuous and/or post-translationally modified epitopes (15); obviously, some epitopes will be too large and/or complex to be included in current peptide microarrays. In terms of discontinuous epitopes, however, it remains to be seen whether a high-density peptide microarray technology will be able to assist in identifying components of discontinuous epitopes. In this context, it is encouraging that others have shown that two low-affinity peptide ligands, when joined, can form a complex high-affinity antibody target (31). In terms of post-translational modifications, whether a particular modified epitope can be generated by our peptide microarray technology depends on whether it is possible to generate the modification in question either during the peptide microarray synthesis or enzymatically after synthesis. A priori, it should be possible to include many modifications (e.g.phosphorylation, glycosylation, etc).
We envision that the location, length, and specificity of linear peptide epitopes conveniently can be identified through a two-step strategy. In the first step, all or most n-mer peptides from the target antigens are synthesized, after which the antibody-binding peptides are selected for synthesis with single-residue substitutions in the second step. A suitable choice of n in the first step seems to be 15, and the offset could be one or a few amino acids. Amino acid scans can then be made in the second step, for which an exhaustive analysis using each of the 20 common amino acids would require at least 1 + 15 × 19 = 286 syntheses for each 15-mer epitope candidate. Our data would suggest that the identification of important residues in a linear epitope can often be obtained from single residue scans made with only one or two amino acids. Thus, an even easier alternative would be to combine a 15-mer length scan with a single amino acid substitution scan. This might enable a simplified “single size fits all” approach. In this case, analyzing a target antigen with a length of 1000 amino acids (about 100 kDa) using 15-mer peptide scans with an offset of one including a single amino acid (say, alanine) scan of each peptide would require the synthesis of some 15,000 peptides. About 75,000 peptides would be needed to generate five copies of each peptide, and such a peptide microarray would still be able to hold all the peptides needed for parallel scans of another 10 similar-sized proteins.
In conclusion, ultrahigh-density peptide microarrays give rise to several advantages over existing methods, including comprehensive coverage of antigens using varying peptide length, short assay time, fast quantifiable fluorescent readout, and streamlined image analysis using tailored software to automatically identify binding regions. Once a polyclonal antiserum has been resolved into distinct peptide epitopes, it should even be possible to use these peptides to affinity purify multiple paired antibody species binding to separate parts of an antigen, thereby allowing one purified antibody preparation to validate the results of another (12). It also paves the way for whole proteome peptide microarrays. Ignoring post-translation modifications, all unique 13-mer peptide sequences in the entire humane proteome can be represented by ∼2,000,000 peptides scanning through the proteome using a peptide length of 18 and overlapping by 12 amino acids. We suggest that a peptide microarray representing the entire humane proteome is within reach.
Acknowledgments
Claus Schafer-Nielsen is the owner and CEO of Schafer-N. Soren Buus, Matthias Uhlén, Johan Rockberg, Björn Forsström, and Peter Nilsson declare no financial interests.
Footnotes
* The research leading to these results has received funding from the European Community’s Seventh Framework Programme ([FP7/2007–2013]) under Grant No. 222773, PepChipOmics.
This article contains supplemental Figs. S1 to S3.
2 Several different padding sequences with amino acids of different natures (negative, positive, hydrophobic, etc.) were tested. In general, the nature of the padding sequence did not affect the results qualitatively (data not shown).
1 The abbreviations used are:
- DCM
- dichloromethane
- DMD
- digital mirror device
- DIEA
- diisopropylethylamine
- Fmoc
- fluorenylmethyloxycarbonyl chloride
- NMM
- N-methylmorpholine
- NMP
- N-methylpyrrolidone
- NPPOC
- 3′-nitrophenylpropyloxycarbonyl
- ANOVA
- one-way analysis of variance
- PrEST
- protein epitope signature tag
- PSSM
- position-specific scoring matrix
- RS
- relative signal
- SSA
- single substitution analysis
- TFA
- trifluoroacetic acid
- LSD
- least significant difference.
REFERENCES
Articles from Molecular & Cellular Proteomics : MCP are provided here courtesy of American Society for Biochemistry and Molecular Biology