![]()
- Published: July 23, 2013
- https://doi.org/10.1371/journal.pone.0068902
Abstract
We have recently developed a high-density photolithographic, peptide array technology with a theoretical upper limit of 2 million different peptides per array of 2 cm2. Here, we have used this to perform complete and exhaustive analyses of linear B cell epitopes of a medium sized protein target using human serum albumin (HSA) as an example. All possible overlapping 15-mers from HSA were synthesized and probed with a commercially available polyclonal rabbit anti-HSA antibody preparation. To allow for identification of even the weakest epitopes and at the same time perform a detailed characterization of key residues involved in antibody binding, the array also included complete single substitution scans (i.e. including each of the 20 common amino acids) at each position of each 15-mer peptide. As specificity controls, all possible 15-mer peptides from bovine serum albumin (BSA) and from rabbit serum albumin (RSA) were included as well. The resulting layout contained more than 200.000 peptide fields and could be synthesized in a single array on a microscope slide. More than 20 linear epitope candidates were identified and characterized at high resolution i.e. identifying which amino acids in which positions were needed, or not needed, for antibody interaction. As expected, moderate cross-reaction with some peptides in BSA was identified whereas no cross-reaction was observed with peptides from RSA. We conclude that high-density peptide microarrays are a very powerful methodology to identify and characterize linear antibody epitopes, and should advance detailed description of individual specificities at the single antibody level as well as serologic analysis at the proteome-wide level.
Figures




Citation: Hansen LB, Buus S, Schafer-Nielsen C (2013) Identification and Mapping of Linear Antibody Epitopes in Human Serum Albumin Using High-Density Peptide Arrays. PLoS ONE 8(7): e68902. https://doi.org/10.1371/journal.pone.0068902
Editor: Morten Nielsen, Technical University of Denmark, Denmark
Received: April 4, 2013; Accepted: June 7, 2013; Published: July 23, 2013
Copyright: © 2013 Hansen et al. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original author and source are credited.
Funding: The research leading to these results has received funding from the European Union Seventh Framework Programme (FP7/2007–2011] under grant agreements 222773 (PepChipOmics) and 278832 (HiPAD). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing interests: The authors have read the journal’s policy and have the following conflicts: Claus Schafer-Nielsen is the owner and CEO of Schafer-N. Lajla Bruntse Hansen and Soren Buus declare no competing interests. This does not alter the authors’ adherence to all the PLOS ONE policies on sharing data and materials.
Introduction
Ideally, the epitope(s) targeted by antibodies used as e.g. diagnostic or therapeutic tools should be identified and extensively characterized in order to validate specificity and to document cross-reactivity that otherwise might lead to spurious results. Unfortunately, current methods of physicochemical epitope characterization tend to be costly, cumbersome, and of low throughput. Examples include X-ray crystallography [1], [2] and multidimensional NMR [3], [4]. As golden standards of epitope characterization these methodologies allow precise identification of the amino acid side chains involved in binding, but they are not suited for large-scale epitope identification and their results cannot be interpreted readily in terms of possible cross-reactions. Other epitope mapping approaches include proteolytic fragmentation [5], analysis of protein arrays and peptide arrays [6], or analysis of recombinant antigen (including antigens arrayed by in situ cell-free translation [7], mutagenized [8] and/or expressed using selectable systems such as phage display [9]). Despite this plethora of epitope-mapping methods, detailed epitope information lacks for the vast majority of antibodies used in life science research. Thus, there is a significant need for comprehensive, yet simple and rapid, methods to map epitopes. Proteins constitute important immune targets and many antibodies used for therapeutic or diagnostic purposes are targeting protein antigens. Traditionally, antibody epitopes in proteins have been classified as being either conformational, i.e. being functional only in spatially constrained forms, or as being linear, i.e. being functional in a form that may be represented by unconstrained peptides [10], [11]. Libraries of linear peptides and of peptides with simple spatial constraints can be produced in various formats and have been used extensively in screenings of antibody epitopes. Early approaches to the synthesis of synthetic peptide libraries involved solid phase synthesis on polystyrene pins (Geysen et al. 1984 [12]), on beads contained in “tea-bags” (Houghten et al. 1985 [13]) or on beads that through a “mix-and-split” strategy (Furka et al. 1991 [14]) allowed the synthesis of “one-bead-one-peptide” libraries (Lam et al. 1991 [15]). While these techniques are able to generate large numbers of different peptides they either require complicated logistics for peptide tracking or they require post-assay sequencing of the positive peptides.
Peptide arrays synthesized and analyzed on planar surfaces simplify the logistics of handling large numbers of peptides and eliminate the need for identification of peptides by sequencing. In a seminal paper, Fodor et al. [16] described the generation of peptide microarrays using a semi-automatic light-directed chemical synthesis of multiple peptides on glass surfaces. Shortly after, Frank et al. [17] described a competing approach involving fully automated synthesis of arrays of peptides with predefined sequences on paper membranes (“SPOT® synthesis”) and this together with the pin-based “PepScan®” method [12] has become a preferred method of generating peptide microarrays. Using SPOT® synthesis, peptide microarray with up to 8000 peptides have been reported [18], and recently high-density peptide microarrays made using a special laser printer technology have been introduced [19]. Light-directed synthesis of peptides has been facilitated by replacement of the physical masks used by Fodor et al. with a digital mirror device (Singh-Gasson et al. [20]) and by introduction of strategies using amino acids with standard protection groups rather than photosensitive groups (Li et al. [21]). Along these lines, we have recently developed a peptide microarray technology that is capable of synthesizing peptide microarrays with up to 2 million pre-addressable peptide fields. Here, we have used this technology to make a complete mapping of linear antibody epitopes in a readily available average sized protein, human serum albumin (HSA) using commercially available polyclonal rabbit anti-HSA antibody as the probe. In a single peptide microarray featuring more than 200.000 peptide fields on a 2 cm2 area, we included 5 copies of all possible 15-mer peptides from HSA. For each of the 595 different 15-mers, we included a complete single-residue substitution analysis with each of the 20 common amino acids and as specificity controls we included all possible 15-mer peptides of bovine serum albumin (BSA) and of rabbit serum albumin (RSA). Using this integrated approach combined with a statistical analysis of the results, we suggest that rabbit anti-HSA antibodies recognize more than 20 linear epitopes and within each of these epitopes we identified the residue positions important for antibody binding together with the amino acid preference of these residues. As expected from the specifications of the manufacturer, the rabbit anti-HSA antibodies showed moderate cross-reactions towards peptides from BSA, while no reaction was observed toward peptides from RSA. We conclude that a systematic overlap and single-residue substitution analysis as enabled by the high-density peptide microarray technology described here allows simultaneous mapping of multiple linear B cell epitopes at the single-residue level.
Methods
Synthesis of High-density Peptide Arrays
Layouts of the arrays were made with proprietary software using the FASTA sequences of HSA (UniProt P02768 Isoform 1), BSA (UniProt P02769) and RSA (RefSeq NP_990592) as input together with definitions of the desired peptide lengths, overlaps, number of copies and amino acids used for single-residue substitutions.
A high-density peptide array was generated using maskless photolithographic synthesis [20]adapted to solid phase peptide synthesis with the C-terminal anchored to the surface [22], [23], [21]. Except for a change in the synthesis substrate (described below) the synthesis was performed as detailed by Schafer-Nielsen and coworkers [24]. Briefly, the image patterns were generated using a.95″ 1920×1080 digital mirror device (DMD, Digital Light Innovations, Austin, Texas) illuminated with collimated 365 nm UV-light. The image patterns generated by 10×10 µm mirrors on the DMD were projected onto the synthesis substrate using 1∶1 UV-imaging optics. The synthesis substrate was a HiSens E microscope slide (Schott AG, Germany) coated by overnight incubation with a 2% w/v linear copolymer of N,N′-dimethylacrylamide and aminoethyl methacrylate (both from Sigma-Aldrich) mixed in a 20∶1 w/w ratio before polymerization for 2 hours at room temperature in freshly degassed 0.1 M sodium borate buffer, pH 8 containing 0.025% v/v TEMED and 0.1% w/v ammonium persulfate. Coupling of amino acids was performed as in classical Fluorenylmethyloxycarbonyl chloride (Fmoc) peptide synthesis using standard Fmoc amino acids (0.1 M amino acid, 0.1 M O-Benzotriazole-N,N,N′,N′-tetramethyl-uronium-hexafluoro-phosphate (HBTU), 0.2 M DIEA in N-methylpyrrolidone (NMP) premixed for 5 minutes before loading into the flow cell). Couplings were performed for 5 minutes after loading of the activated amino acid. The principal deviation from the classical strategy was that the photolabile (2-(2-nitrophenyl)propyl oxycarbonyl (NPPOC) was coupled to amino groups on the surface of the substrate before onset of synthesis, and when all n-terminal amino groups in a given layer had been coupled and the Fmoc-groups had been removed with 20% v/v piperidine in NMP for 20 minutes. Couplings of NPPOC were made by incubation for 30 min with a 1% v/v solution of NPPOC-chloroformate (Sigma), 0.1 M DIEA in DCM/NMP 1∶4 v/v. Before coupling with a new amino acid, relevant areas of the synthesis substrate were irradiated in 0.1 M DIEA in NMP for 10 minutes with 365 nm UV-light at an energy density of 20 mW/cm2. All synthesis steps were performed at room temperature by a proprietary liquid handling robot connected to a locally constructed DMD-projection device equipped with a flow cell holding the synthesis substrates. After synthesis, side chain protecting groups of peptides in the array were cleaved by incubation in Trifluoroacetic acid (TFA): 1,2-ethanedithiol (EDT): Triisopropylsilane (TIPS):H2O, 92∶2:1∶5 v/v/v/v overnight at room temperature before washing in DCM and air drying.
Binding, Staining and Recording of the Array
The microscope slide with the high-density peptide array was incubated for 2 hours in polyclonal rabbit anti-human HSA (A001, DAKO) diluted 1+100 in 0.15 M Tris (Trizma® base, Sigma-Aldrich)/acetate pH 8.0, 0.1% v/v Tween20 (dilution and washing buffer). After washing, the slide was incubated for 2 hours in a 1+1000 dilution of Cy3-conjugated goat anti-rabbit IgG (A10520, InVitrogen) followed by repeated washings. Finally, the array was visualized and recorded using a MVX10 fluorescence microscope (Olympus) equipped with a MX10 cooled camera connected to a computer with CellP© imaging software (Olympus).
Analysis of the Array Images
Digital images of the array were scanned and analyzed by proprietary software. The results were stored in text files as TAB-delimited lists including the sequences in each peptide field and the average fluorescence intensity recorded in that field. Background correction was performed by subtraction of the intensity recorded in nearby “blank” fields (i.e. fields with no peptide, only linker residues). The signal in the outer 30% of the rim of each field was discarded to minimize carryover of signals between the fields.
Statistical Analysis
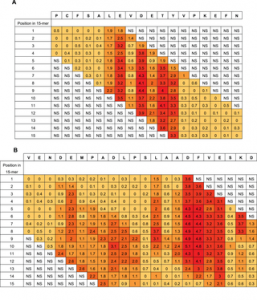
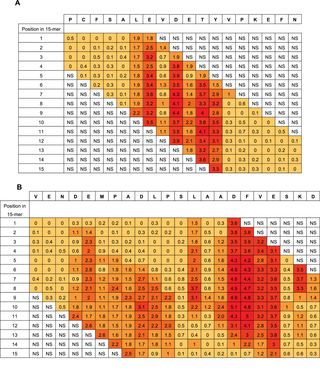
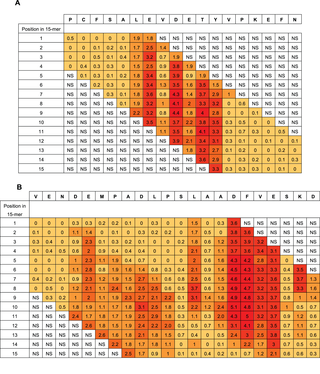
A complete single-residue scan was performed for each 15-mer peptide from HSA, and the signal obtained for each substituted peptide was subsequently expressed in percentage to the signal of the corresponding native 15-mer peptide. The normalized values were arranged in position specific scoring matrices (PSSM) with 15 columns representing the positions of the native peptide and 20 rows representing the 20 naturally occurring amino acids (Figure S2). An arithmetic mean of the normalized values, µ, was calculated for each position. Each PSSM was subjected to an analysis of variance (ANOVA) followed by a post-hoc analysis using Tukey’s Honest Significance Difference (HSD). The HSD value was used to identify positions where µ differed significantly from 100% and thus, contribute to the selectivity of the antibody-peptide interaction at a chosen significance level. HSD is calculated according to the formulaWhere qcrit is the relevant critical value of the studentized range, i.e. the value found in q-tables corresponding to the available degrees of freedom at the chosen level of significance (p<0.01). MSwithin is the mean square error and n is the number of substitutions used in calculation of the mean values.
The obtained value of the studentized range (qobt) is calculated as:
The obtained q-value is compared with the critical q-value by calculating the ratio (Rq) between these two values:
The ratio will be ≥1.0 for all positions with residues that contribute to the binding of antibody.
Results
Analysis of signals from a high-density array probed with anti-HSA antibodies
A microarray with more than 200.000 peptide fields was designed and synthesized as described in the method section. In addition to 15-mer peptides derived from the native sequences of HSA, BSA and RSA, the array contained 300 single-residue substituted variants of each of the 595 different 15-mers from HSA, and more than 20.000 reference peptide fields. The protein sequences of HSA, BSA and RSA are listed in Figure S1.
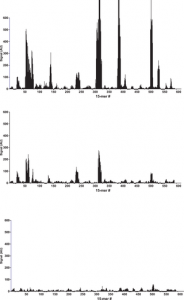
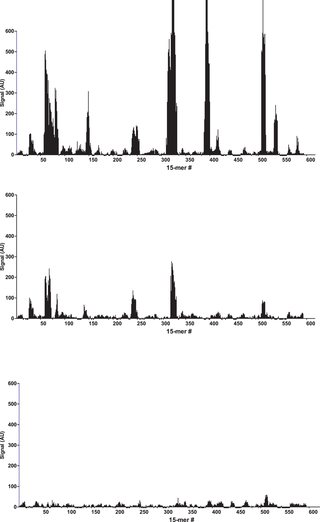
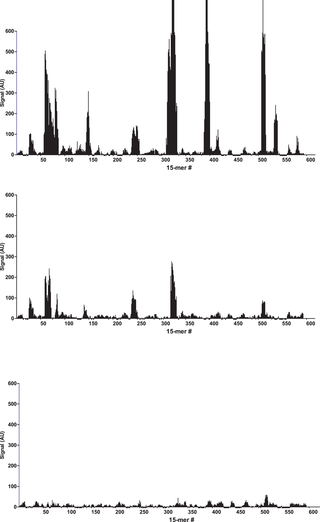
After binding of the antibody, images of the array (Fig. 1) recorded using fluorescence microscopy revealed discrete fluorescence of sharply demarcated quadratic fields consistent with a successful optical projection of the digital mirrors onto the peptide microarray. A plot of the signal intensities obtained from the overlapping 15-mer peptides in HSA (Fig. 2A) shows clear accumulation of signals corresponding to discrete regions of the HSA sequence. Both the width and the height of the signal peaks differ from one region to the next and depending on the criteria used for inclusion it can be readily estimated that 10–20 signal peaks are discernible.