
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Fig. 4.
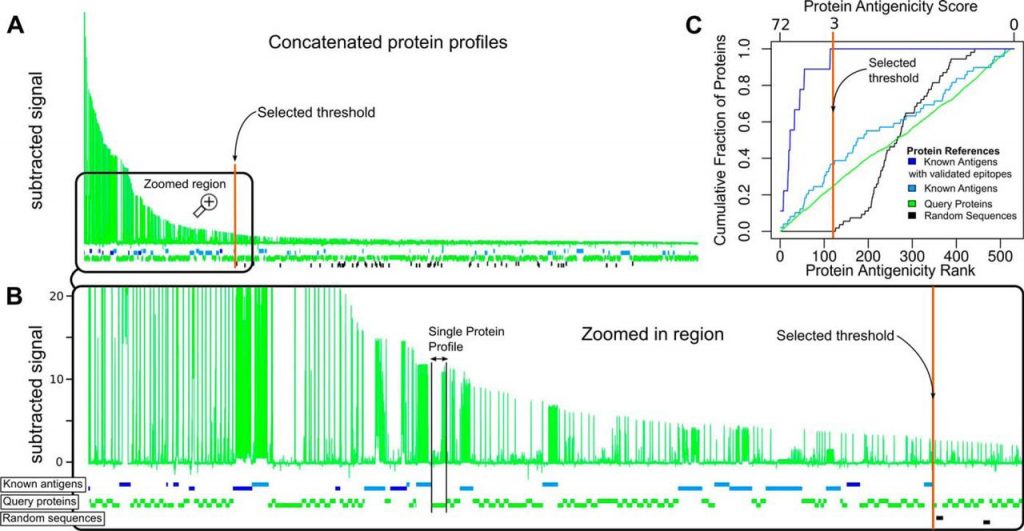
Global visualization of antigen and epitope discovery. A, Concatenation of all protein profiles (similar to those in Fig. 1) ordered from left to right according to the peptide with the highest signal in each protein. The x axis shows the residue position for all concatenated proteins (full length including their antigenic and non-reactive regions), and the y axis represents the averaged, normalized and subtracted reactivity values (Chagas-specific signal). B, Enlarged region showing the antigenicity profiles of proteins that lie to the left of the selected threshold value of 3 (colored vertical line). Horizontal boxes intercalated below the concatenated profiles represent individual proteins. Blue boxes represent antigens with previously validated linear epitopes. Cyan boxes are proteins with previous serology evidence (Protein Group 4) but without known linear epitopes. Black boxes correspond to random sequences (Group 5). Green boxes represent serologically uncharacterized (‘query’) T. cruzi proteins (Groups 1, 2 and 3). C, Enrichment of different groups of proteins along the ranking. Bottom x-axis: antigenicity rank, with the most antigenic protein (according to the highest scoring peptide) at the left. Top x-axis: protein antigenicity score. In the y-axis, the cumulated fraction of proteins at each ranking position, for the different protein group.
Using this threshold, around 1% of all peptides were hence found positive with substantial positivity rate differences across protein groups. Group 4 had the highest positivity rate with ∼2.62% of its peptides being positive, followed by Group 3 of MASP family proteins with 1.21% of positive peptides, followed by Group 2 containing peptides from prioritized proteins with a rate of 0.84%; and finally Group 1 (randomly selected proteins) with a 0.13% rate. Noteworthy, our integrative multi-feature prioritization strategy (25) produced a ∼sixfold increment in the peptide reactivity rate compared with selecting proteins at random (group 2 versusgroup 1). Detailed reactivity rates for each protein group are summarized in Table I, and protein profiles for all positive proteins are shown in supplemental Fig. S2.
Table IChagas-specific positivity rates in different peptide sets. The table shows the total number of distinct peptides assayed in each group; the number of positive peptides (above cutoff of 3) in the averaged signal (combining data from eight peptide-chip assays); the average number of positive peptides in a single experiment (single chip/single sample above cut-off of 7); and the number of peptides that were positive in at least 1 out of the 4 sera samples (pools) tested. Percentages are given in parentheses. Group 5 of 24,000 randomly generated peptides (negative control) had no positives in any case
Next, to further delineate antigenic portions of the analyzed proteins we defined any group of consecutive reactive peptides located in a specific region of an antigen as an ‘antigenic region’ (see “Experimental Procedures”). These regions are defined by peaks in the antibody-binding signal observed from the arrays and contain one or more B-cell epitopes. The 2031 positive peptides detected were mapped to 187 distinct antigenic regions. After removal of peptides from known T. cruzi antigens, the remaining 1164 peptides (121 unique regions, which map to 97 novel T. cruzi antigens), represent new peptidic markers for Chagas Disease, for which this report represents their first serological characterization. Furthermore, from the 24 positive proteins with prior serologic evidence (Group 4), we detected new antigenic regions (not reported previously) in 10 of them (shown in supplemental Table S2). Protein reactivity rates for each protein group are summarized in Table II. Briefly, 35% of proteins in Group 4 were Positive, followed by a 30% of Group 3 (MASPs), followed by a 21% of prioritized protein (Group 2) and a 10% of randomly picked proteins (Group 1).
Table IIChagas-specific positivity rates in different protein sets. The table shows the total number of distinct proteins assayed; the number of proteins with any specific signal peak above the specified cutoff considering the averaged signal (combining data from eight peptide-chip assays); the average number of positive proteins in a single experiment (single chip\sample); and the number of proteins that displayed a positive signal (above cutoff) in any of the four sera samples tested. Percentages are given in parentheses. Group 5 of randomly generated 54 neo proteins (negative controls) had no positives in any case
In summary, our study is providing detailed information on the linear antigenic determinants for both novel antigens, and for a subset of antigens that have not been characterized in detail until now. A table with all antigenic regions along with their source protein, localization, sequence and reactivity data is available as supplemental Table S2.
It is worth mentioning that prior to this work, the number of known T. cruzi antigens recognized from human infections was ∼68, and the number of those with mapped epitopes were 20 (see supplemental Table S3). Therefore, despite the fact that our HD-Chip contained a still low representation of the T. cruzi proteome (∼3% of the proteome was displayed in our design), we were able to increase the coverage of known antigens for this pathogen more than twofold, and more than 10-fold in the case of finely mapped antigenic regions, essentially providing the majority of currently known epitopes for this disease.
DISCUSSION
This study provides so far the largest number of fine antibody specificities simultaneously measured for a human infection, with more than a 10-fold increment in throughput when compared with previous screenings using peptide microarrays (7, 13, 42, 43). With only 20 μg of purified immunoglobulin directly obtained from human clinical samples, these high-density peptide microarrays allowed the recognition of specificities against ∼180,000 distinct pathogen-specific 15-mer peptides. Moreover, a sequential incubation protocol allowed subtraction of signal from healthy/non-infected human samples to obtain infection-specific signal in each microarray slide.
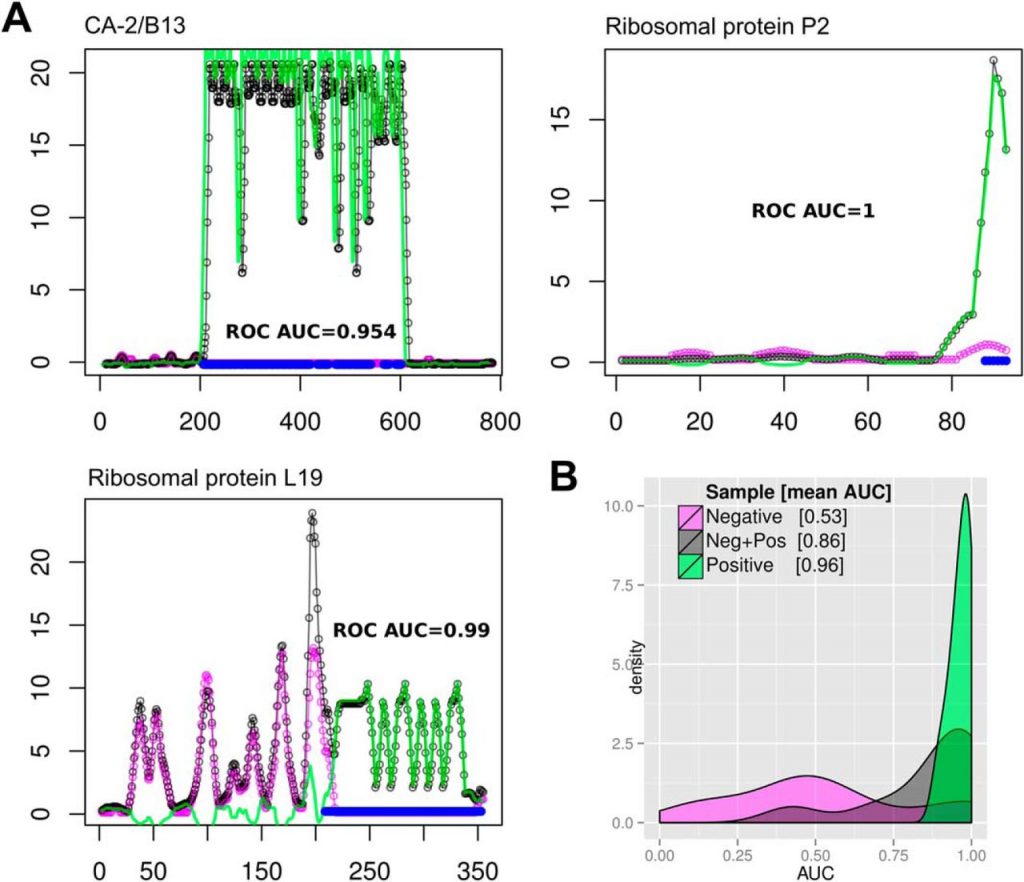
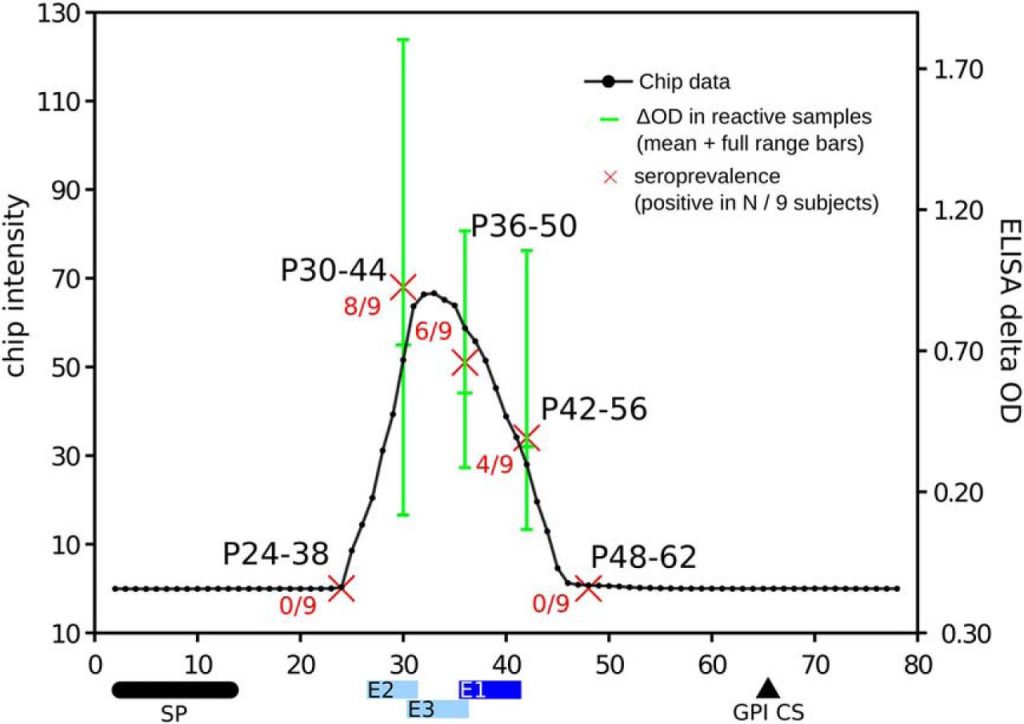
We benchmarked the precision of our measurements using previously known antigens with mapped B-cell linear epitopes. These experiments showed that HD-Chips have an excellent epitope mapping performance, with AUCs values >0.96 in a single microarray and an average AUC of 0.972 after combining data from multiple samples. Further analysis showed that the deviation from a perfect AUC of 1 was only because of single-residue differences at the boundaries of the previously reported B-cell epitopes, particularly in the case of antigens with multiple repetitive epitopes. An independent validation in ELISA format of the TSSA antigen profile further supported the mapping precision of HD-Chips. In fact, we showed in this case that the major linear epitope within this antigen is located between residues 30 and 46 and not between residues 41 to 50 as detected using a less accurate technique (39).
The set of 15-mer peptides derived from randomly generated protein sequences was essential to define a nonspecific antibody-binding baseline distribution, and to normalize the data, allowing the subtraction of signal from incubations with negative samples, and bringing the data from different microarray replicates into a common scale. These data was also used to guide the selection of a reasonable threshold for classification of reactive peptides. Using this threshold, more than 2000 peptides, i.e. ∼1% of all screened 15-mer peptides, resulted seropositive in T. cruzi infected human sera pools and negative in the non-infected sera pools, with virtually no false positives (with the reactivity of all 24,000 randomly generated peptides below the cut-off). Although this threshold was defined based on the comparison against the reactivity of random peptides, it is already well established that both combinatorial libraries in phages (1), or random peptides in high-density arrays (43, 44) can detect significant and reproducible binding by mimicking natural epitopes. Therefore, although we chose a very conservative threshold to identify the most reactive peptides, lowering this threshold can certainly provide additional Chagas-specific biomarkers.
In our benchmark, epitope mapping performance was enhanced by subtracting reactivity signals measured in healthy donor samples, i.e. not related to the infection of interest, and perhaps caused by exposure of patients to other infectious agents or antigens with cross-reacting epitopes. Moreover, with this experimental setup the two samples were compared on the same physical spots, hence reducing the number of required chips by half and suppressing the variability associated with peptide synthesis and slide manipulation.
Based on the presented evidence it is clear that these HD-Chips are highly suitable for exhaustive mapping of the specificities of B-cell responses against human pathogens. Interestingly, antibody-binding to specific pathogen protein regions was also detected when using sera pools from healthy individuals (see for example the N-terminal region of the Ribosomal protein L19 in Fig. 1A, additional cases are also evident in supplemental Fig. S2). These antigenic determinants may be shared with other pathogens to which the donor has been exposed, or may represent cross-reactive determinants to other antigens (perhaps from vaccination antigens). In any case, the high-resolution scanning of antibody-binding across the full length of proteins is able to precisely map these regions, providing essential information for improving the specificity of recombinant antigens used in diagnostic applications.
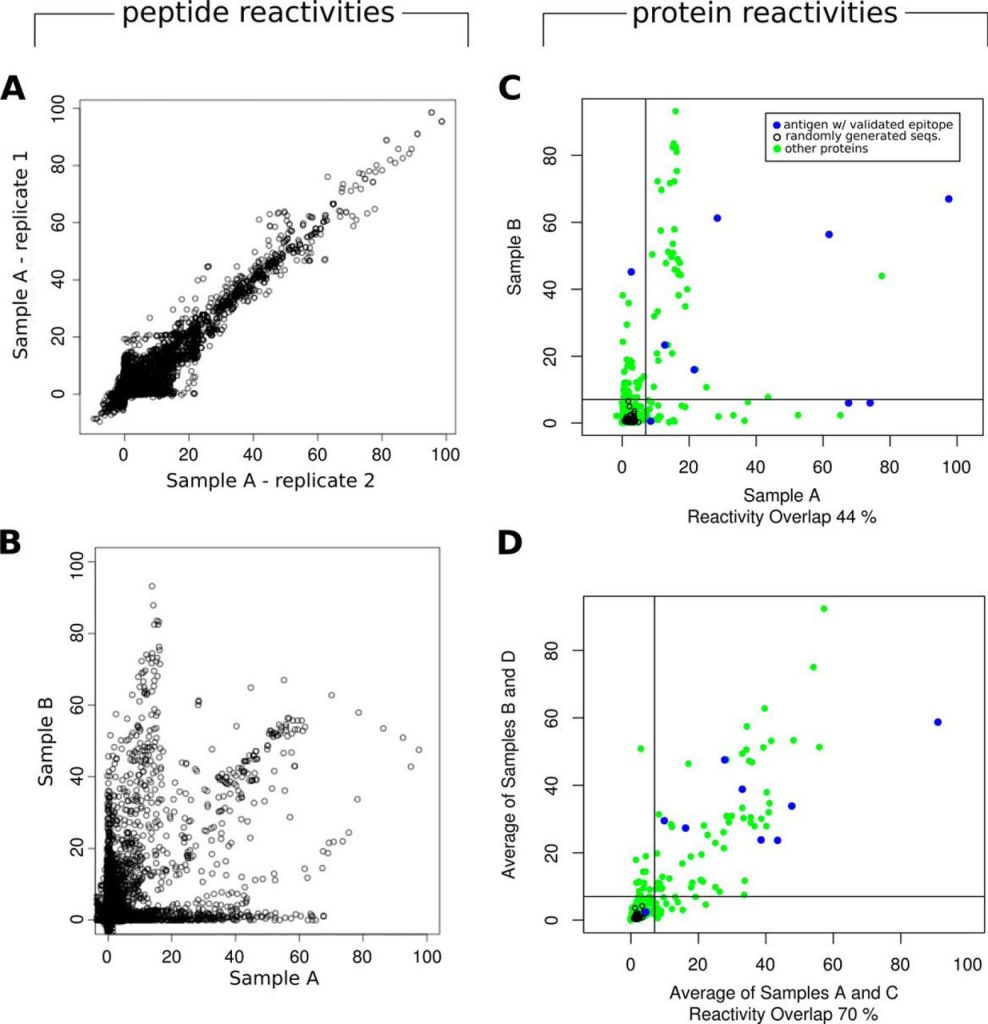
In this context, it is interesting to compare the overall rate of antigen discovery provided by peptide and whole-protein arrays. Previous studies have profiled humoral immune responses in infected humans compared with healthy individuals for several pathogens using protein microarrays (45). Some of the largest studies were performed for Mycobacterium tuberculosis (∼4000 proteins, representing almost the full proteome; with a 10% of seroreactive proteins) (8), Leptospira interrogans (∼3300 proteins, representing almost full proteome; with ∼5% of seroreactive proteins) (46), Brucella melitensis (∼3000 proteins, representing almost full proteome, with a ∼4% of seroreactive proteins) (47), and Plasmodium falciparum (∼1200 proteins or ∼23% of its proteome, with ∼13% of seroreactive proteins) (9). In our experiments we obtained a ∼1% global peptide positivity rate (∼2,031 positive out of 175,566 unique T. cruzi derived 15-mer peptides) when averaging signal from all HD-Chip experiments. This rate, goes down to ∼0.5% if we consider a non-redundant set of proteins with no prior serological characterization. In terms of full-length proteins, this corresponds to a ∼15% of the parasite proteins being targeted by the humoral immune response in infected subjects. Despite the fact that peptide microarrays are limited to identify linear epitopes, the estimated proportion of seroreactive proteins observed here for Chagas Disease is in line and even above those described in the aforementioned screenings using whole protein microarrays in other pathogens.
Therefore, we believe that high-density peptide microarrays offer an excellent platform for whole proteome screenings. In scenarios where the peptide space to screen is still too large, a combination of high-coverage whole-protein arrays followed by a detailed epitope scanning using high-density peptide chips could provide an excellent strategy to analyze antibody responses against pathogens. In this strategy, protein arrays would streamline the process of antigen identification, and HD peptide chips would precisely map the antigenic determinants in these antigens.
Besides the obvious practical impact of these large scale antigen screenings on the development of improved serodiagnostics and vaccines, unbiased and exhaustive studies of humoral immune response specificities in human infectious diseases are needed for a better understanding of pathogen immunogenicity and immunodominance, and to improve current antigenicity prediction tools (48, 49). In our study, we successfully detected all antigenic determinants in high-confidence antigens with mapped B-cell epitopes. We were also able to discover and map the locations of epitopes for previously described antigens that lacked detailed B-cell epitope mappings. However, we failed to detect linear antigenic determinants for some of the proteins with previous serological evidence. Among other likely explanations, these antigens may contain only discontinuous, or low prevalence epitopes. Low prevalence of epitopes may be explained by the genetic heterogeneity of infecting parasites (50). Future studies using diverse serum samples (e.g. from diverse geographic origins, or from patients displaying different clinical manifestations of the disease) will help to answer this. We also note that the only epitopes that are readily mimicked by synthetic peptides are continuous epitopes. Discontinuous epitopes, which are made up of residues from separate stretches of the antigen polypeptide chain, can only be identified by x-ray crystallography of antigen-antibody complexes. However, we believe this limitation is largely balanced by the throughput and flexibility offered by high-density peptide microarrays.
For many of these screenings seeking to identify antigens or defined antigenic determinants, a number of bioinformatic strategies based on defined protein features have been applied to narrow down the set of candidates displayed in the arrays (25, 51⇓–53). We have previously developed one such strategy to prioritize T. cruzi proteins and peptides for use in a spotted peptide microarray (25). In this work we found a significant ∼6 fold increment in the number of seroreactive peptides in this ranked set when compared with a random pick, thus supporting the utility of such approaches.
In these experiments, we used a HD-Chip design were we simultaneously measured antibody-binding to >200,000 addressable spots in each slide. However, larger capacities (currently up to two million addressable spots) are possible by using a single mirror per peptide field. In this case, the larger capacity comes at the price of smaller fields and inherent larger measurement error. However, even using larger peptides fields, the screened protein space can be increased significantly by decreasing the sequence overlap between adjacent peptides in a protein sequence. The high signal correlation observed from peptides that are contiguous in the protein sequence suggested that it could be possible to further reduce the number of peptides required to completely scan each protein. In a posteriori computational experiments in which we re-analyzed the data simulating different peptide overlaps, we observed no significant epitope mapping performance loss down to an overlap of 12 residues between two 15-mers (i.e. a 3 residue shift when scanning a protein sequence) (not shown). This means that we should be able to achieve a ∼threefold increase in the protein search space using the same number of peptides per slide. Considering a large proteome such as that of T. cruzi, 33 HD-Chips would be required to screen the ∼6 million 15-mers in which this proteome can be broken down (200K peptide fields per chip, scanning proteins at maximal resolution). However, by scanning proteins with a 12-residue overlap and pushing the microarray density up to 500K peptides per slide, the complete proteome could be covered with only four HD-Chips, without sacrificing significant sensitivity.
In conclusion, by taking advantage of next-generation peptide arrays, we show that by screening ∼3% a large eukaryotic proteome we discovered and finely mapped more than 120 new antigenic determinants, providing essentially most of the linear B-cell epitopes currently known for this infectious disease. Our results show that it is now feasible to increase the pace of biomarker discovery for infectious diseases, and to further increase the scale and detail in the study of B-cell immune responses against human infectious diseases.
Acknowledgments
We thank Dr. Nick Thomson (Wellcome Trust Sanger Institute, Cambridge, UK) for critical reading of the manuscript, and Lic. María Gabriela Figini (IIB-INTECH, UNSAM) for technical support.
Footnotes
Author contributions: S.J.C. and F.A. designed research; S.J.C., C.S., V.B., C.A.B., and F.A. performed research; C.S. and J.A. contributed new reagents or analytic tools; S.J.C., M.N., J.M., V.T., A.C.F., O.C., C.A.B., and F.A. analyzed data; S.J.C., M.N., C.A.B., and F.A. wrote the paper.
↵* This work was funded by grants from the Agencia Nacional de Promoción Científica y Tecnológica, Argentina (FITS-Chagas-003, PICT-2013-1193), and from the European Union Seventh Framework Programme (FP7/2007-2011) under grant agreements 222773 (PepChipOmics) and 278832 (HiPAD). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
↵
 This article contains supplemental Figs S1 to S3 and Tables S1 to S5.
This article contains supplemental Figs S1 to S3 and Tables S1 to S5.DISCLOSURE DECLARATION. Claus Schafer-Nielsen is the owner and CEO of Schafer-N ApS. The other authors declare no conflict of interest.
↵1 The abbreviations used are:
- HD-Chips
- high-density peptide chips
- ROC
- Receiver Operating Characteristic
- AUC
- Area under the ROC curve
- PPV
- Positive-Predictive Value
- PCC
- Pearson product-moment Correlation Coefficient.
- Received October 27, 2014.
- Revision received March 26, 2015.
- © 2015 by The American Society for Biochemistry and Molecular Biology, Inc.
Author’s Choice—Final version free via Creative Commons CC-BY license.